Das AMNOG-Verfahren
Seit dem Inkrafttreten des Arzneimittelmarktneuordnungsgesetzes (AMNOG) am 1. Januar 2011 bewertet der Gemeinsame Bundesausschuss (G-BA) den Nutzen von neuen Arzneimitteln mit in der Regel neuen Wirkstoffen. Zentrale Bausteine sind die Bewertung des Zusatznutzens gegenüber der zweckmäßigen Vergleichstherapie (zVT), die Bestimmung des Ausmaßes des Zusatznutzens und dessen therapeutische Bedeutung. Der daraus resultierende Nutzenbewertungsbeschluss des G-BA bildet die Basis für die Verhandlung des sog. Erstattungsbetrags zwischen GKV-Spitzenverband und dem Arzneimittel-Hersteller.
Das AMNOG trat mit folgenden Zielen1 in Kraft:
- Den Menschen müssen im Krankheitsfall die besten und wirksamsten Arzneimittel zur Verfügung stehen.
- Die Preise und Verordnungen von Arzneimitteln müssen wirtschaftlich und kosteneffizient sein.
- Es müssen verlässliche Rahmenbedingungen für Innovationen, die Versorgung der Versicherten und die Sicherung von Arbeitsplätzen geschaffen werden.
Demnach ist eine Balance zwischen Innovation und Bezahlbarkeit von neuen Arzneimitteln mit einer stärkeren Orientierung am Wohl der Patientinnen und Patienten zu finden. Der Zusatznutzen der Arzneimittel für die Patientinnen und Patienten bestimmt seitdem den Preis neuer Arzneimittel.
1 Deutscher Bundestag, Drucksache 17/2413, 17. Wahlperiode, 06. 07. 2010.
[glossar] Zweistufiges Prinzip des AMNOG-Verfahrens :::
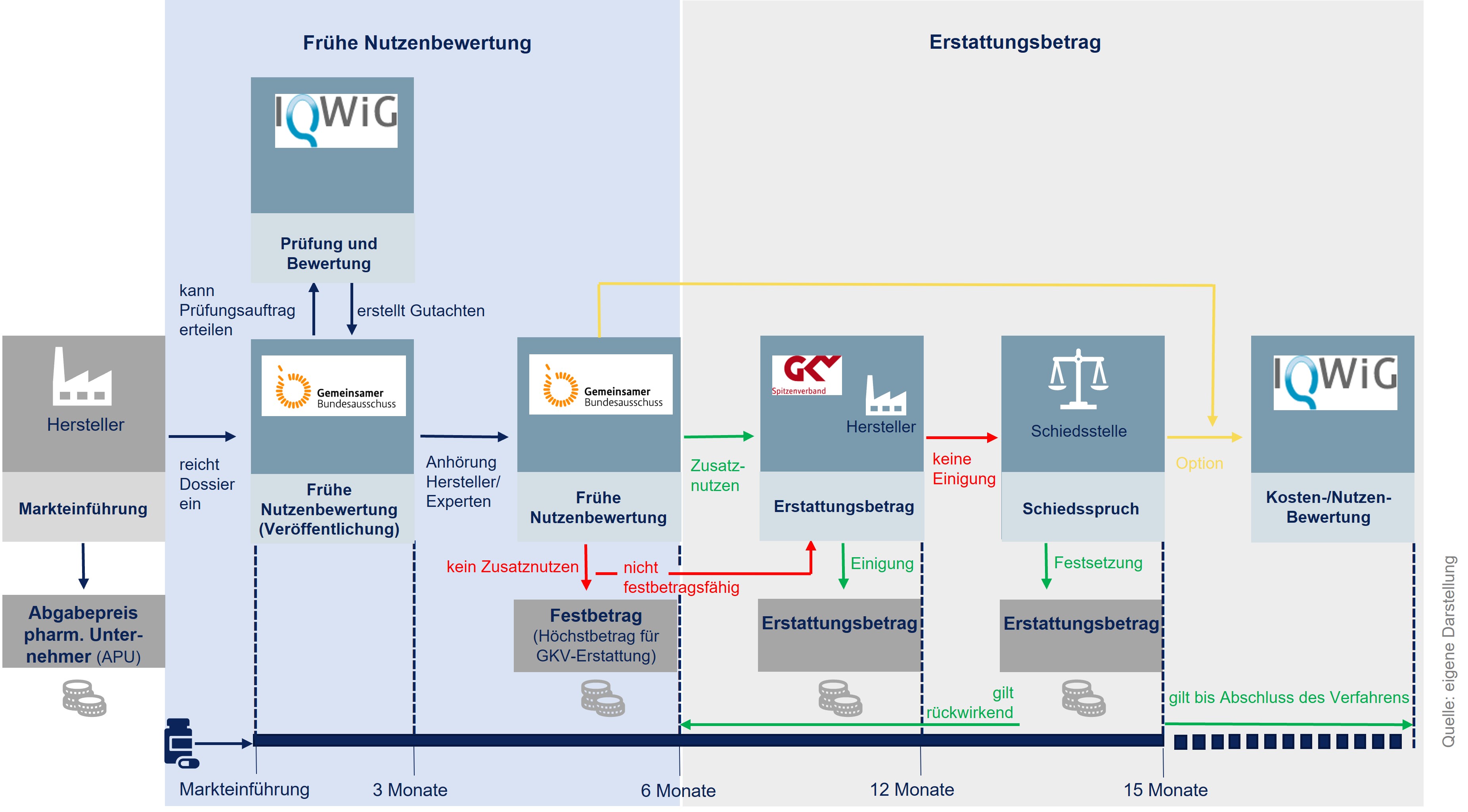
Das AMNOG-Verfahren beginnt unmittelbar mit Markteinführung und folgt einem zweistufigen Prinzip (siehe Abbildung 1). Im Rahmen der frühen Nutzenbewertung bewertet der G-BA zunächst im ersten Schritt des Verfahrens den Zusatznutzen gegenüber einer etablierten Therapieoption (=zweckmäßige Vergleichstherapie, kurz: zVT) nach Maßstäben der Evidenzbasierten Medizin. Hierzu kann er das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) beauftragen. Im zweiten Schritt finden daraufhin Verhandlungen zwischen dem Arzneimittel-Hersteller und dem GKV-Spitzenverband über den Erstattungsbetrag statt. Das gesamte Verfahren ist in der Regel nach einem Jahr abgeschlossen. Mit Inkrafttreten des GKV-Finanzstabilisierungsgesetzes am 12. November 2022 gilt dieser Erstattungsbetrag rückwirkend ab dem siebten Monat nach dem erstmaligen Inverkehrbringen des Wirkstoffs statt des bis dahin geltenden durch den Hersteller festgelegten Preises. [/glossar]
[glossar] Frühe Nutzenbewertung :::
Arzneimittel mit neuen Wirkstoffen werden nach ihrer Zulassung einer frühen Nutzenbewertung durch den G-BA unterzogen. Dies gilt auch für Arzneimittel mit neu zugelassenen Anwendungsgebieten, sofern diese nach dem 1. Januar 2011 erstmals zugelassen wurden. Das Sozialgesetzbuch V (SGB V) und die Verfahrensordnung des G-BA definieren weitere Bewertungsfälle.
Im Gegensatz zur Zulassung eines Arzneimittels, bei der die Qualität, Unbedenklichkeit und Wirksamkeit eines neuen Arzneimittels und damit dessen Nutzen geprüft wird, prüft die frühe Nutzenbewertung die patientenrelevanten Effekte im Vergleich zu bereits vorhanden Therapieoptionen und damit den Zusatznutzen. Entscheidend dabei ist, ob bei dieser Prüfung ein Zusatznutzen gegenüber der sogenannten zVT identifiziert werden kann. Der Zusatznutzen eines Arzneimittels ist nach den Vorschriften der Arzneimittel-Nutzenbewertungsverordnung ein Nutzen, der quantitativ oder qualitativ höher ist als der Nutzen, den die zVT aufweist.
Bei der Bewertung des Zusatznutzens wird zwischen Ergebnissicherheit und Ausmaß unterschieden.
Die Ergebnissicherheit erlaubt eine Einschätzung darüber, inwieweit das vorhandene Wissen, in diesem Fall die relevanten Studienergebnisse, zuverlässig sind. Details, wie Studienplanung, -ausführung, -auswertung und -veröffentlichung haben direkten Einfluss auf die Verlässlichkeit der Studienergebnisse. Auf Basis dieser Evidenz kann somit entweder ein Anhaltspunkt, ein Hinweis oder ein Beleg abgeleitet werden.
Das Ausmaß des Zusatznutzens beschreibt hingegen den Therapieerfolg eines Arzneimittels gemäß den in Tabelle 1 dargestellten Kategorien der Arzneimittel-Nutzenbewertungsverordnung.
Tabelle 1: Kategorien nach Arzneimittel-Nutzenbewertungsverordnung
| Ausmaß | Definition |
|---|---|
| erheblicher Zusatznutzen | Nachhaltige und gegenüber der zweckmäßige Vergleichstherapie (zVT) bisher nicht erreichte große Verbesserung des therapierelevanten Nutzens, insbesondere Heilung der Erkrankung, erhebliche Verlängerung der Überlebensdauer, eine langfristige Freiheit von schwerwiegenden Symptomen oder die weitgehende Vermeidung schwerwiegender Nebenwirkungen |
| beträchtlicher Zusatznutzen | Gegenüber der zVT bisher nicht erreichte deutliche Verbesserung des therapierelevanten Nutzens, insbesondere eine Abschwächung schwerwiegender Symptome, eine moderate Verlängerung der Lebensdauer, eine spürbare Linderung der Erkrankung, eine relevante Vermeidung schwerwiegender Nebenwirkungen oder eine bedeutsame Vermeidung anderer Nebenwirkungen |
| geringer Zusatznutzen | Gegenüber der zVT bisher nicht erreichte moderate und nicht nur geringfügige Verbesserung des therapierelevanten Nutzens, insbesondere eine Verringerung von nicht schwerwiegenden Symptomen der Erkrankung oder eine relevante Vermeidung von Nebenwirkungen |
| Zusatznutzen nicht quantifizierbar | wissenschaftliche Datenlage lässt eine Quantifizierung des Zusatznutzens nicht zu |
| Zusatznutzen nicht belegt | kein Zusatznutzen belegt |
| geringerer Zusatznutzen | geringerer Nutzen gegenüber der zVT |
Für die Bewertung des Zusatznutzens hat der Arzneimittel-Hersteller mit der Markteinführung ein Dossier auf Basis der eigens durchgeführten klinischen Studien oder als relevant identifizierten klinischen Studien Dritter einzureichen. Das Dossier enthält alle relevanten Angaben über Kosten und patientenrelevanten Zusatznutzen des Arzneimittels im Vergleich zur zVT. Auf dieser Grundlage entscheidet der G-BA innerhalb von drei Monaten, ob das Arzneimittel gegenüber der zuvor festgelegten zVT einen medizinischen Zusatznutzen aufweist oder nicht. Für diese Prüfung kann der G-BA das IQWiG oder Dritte beauftragen. [/glossar]
[glossar] Verhandlung über Erstattungsbetrag :::
Das Ergebnis der frühen Nutzenbewertung führt zu folgenden möglichen Situationen:
- Arzneimittel ohne Zusatznutzen und mit Festbetragsgruppe
Wird kein Zusatznutzen gegenüber der Vergleichstherapie festgestellt, dann wird das Arzneimittel einer Festbetragsgruppe zugeordnet, sofern diese bereits besteht und das Arzneimittel dieser zugeordnet werden kann.
- Arzneimittel ohne Zusatznutzen und ohne Festbetragsgruppe
Wird kein Zusatznutzen gegenüber der Vergleichstherapie festgestellt und das Arzneimittel kann keiner Festbetragsgruppe zugeordnet werden, verhandeln GKV-Spitzenverband und Arzneimittel-Hersteller einen Erstattungsbetrag. Dabei ist zu berücksichtigen, ob die zVT noch Patent- oder Unterlagenschutz genießt oder nicht. Bei mehreren zVT ist zudem auf die Wirtschaftlichste abzustellen.
- Arzneimittel mit Zusatznutzen
Wird ein Zusatznutzen gegenüber der Vergleichstherapie festgestellt, wird in Abhängigkeit seines Ausmaßes und der Wahrscheinlichkeit ein Erstattungsbetrag verhandelt. Im Falle eines nicht quantifizierbaren oder geringer Zusatznutzens und wenn zugleich die zVT noch einem Patent- oder Unterlagenschutz unterliegt, dürfen die Jahrestherapiekosten des neuen Arzneimittels nicht höher als die der zVT sein. Der Erstattungspreis für Arzneimittel mit einem beträchtlichen oder erheblichen Zusatznutzen soll den Zusatznutzen der Innovation angemessen reflektieren.
Kommt es zwischen GKV-Spitzenverband und Arzneimittel-Hersteller zu keiner Einigung, wird eine Schiedsstelle angerufen, die den Erstattungsbetrag per Schiedsspruch festlegt. Der Erstattungsbetrag gilt dann rückwirkend ab dem 7. Monat des Inverkehrbringens. Wird der Schiedsspruch nicht akzeptiert, kann er vor dem Landessozialgericht Berlin-Brandenburg beklagt werden. [/glossar]
[glossar] European Health Technology Assessment (EU-HTA) :::
Ab Januar 2025 wird diesem rein nationalen Verfahren ein harmonisierter europäischer Prozess (EU-HTA) vorausgehen, sofern für das entsprechende Arzneimittel eine europäische Zulassung besteht. Im EU-HTA Verfahren soll bereits die wissenschaftliche Evidenz der vorgelegten Studiendaten überprüft werden, ohne jedoch eine Wertung der Ergebnisse vorwegzunehmen. Nähre Informationen finden Sie im Bereich Unsere Themen > Arzneimittel in Europa > Europäischer HTA-Prozess auf unserer Webseite. [/glossar]
Zukunftsorientierte Datennutzung im AMNOG-Prozess
Pharma Deutschland hat dazu drei zentrale Ansätze formuliert:
Eine einheitliche Datengrundlage für beide Verhandlungsparteien ist essenziell für faire und ausgewogene Verhandlungen. Eine exklusive Datenhoheit des GKV-Spitzenverbandes würde das Gleichgewicht im Verhandlungsprozess beeinträchtigen und ist daher abzulehnen. Ein gleicher Zugang zu allen relevanten Daten stellt sicher, dass Entscheidungen auf einer objektiven und gemeinsamen Basis getroffen werden können. Dies dient nicht nur den Interessen aller Verhandlungspartner, sondern kommt letztlich auch den Patientinnen und Patienten zugute.
Zügige Antragsbearbeitung beim Forschungsdatenzentrum: Für eine effektive Nutzung der Daten ist aus Sicht des Verbandes eine schnelle und unbürokratische Bearbeitung von Anträgen unerlässlich.
Die Berücksichtigung versorgungsnaher Daten ist von zentraler Bedeutung, da diese klinische Studien ergänzen und eine fundierte Beurteilung des tatsächlichen Nutzens von Arzneimitteln ermöglichen. Dies trägt zur Verbesserung der Datenqualität bei, unterstützt Entscheidungsprozesse und fördert Innovationen im Gesundheitswesen. Versorgungsnahe Daten stellen einen wesentlichen Bestandteil bei der Bewertung neuer Therapien dar und sollten stärker in die Nutzenbewertung einbezogen werden. Gleichzeitig muss die Methodik zur Bewertung dieser Daten an die neuen Zugangsmöglichkeiten angepasst und weiterentwickelt werden.