Europäischer HTA-Prozess

Am 12. Januar 2025 beginnt eine neue Epoche in der Bewertung des Nutzens von Arzneimitteln. Ab diesem Datum gilt die Europäische Health Technology Assessment Verordnung (EU-HTA). Schrittweise wird die Bewertung von Gesundheitstechnologien für Arzneimittel, Medizinprodukte und In-vitro-Diagnostika (nur für spezifische Klassen) eingeführt. Für neue Arzneimittel ist künftig ein separater Bewertungsprozess (Health Technology Assessment, kurz: HTA) neben dem Zulassungsverfahren verpflichtend.
[glossar] Warum werden Health Technology Assessments (HTA) durchgeführt? :::
Arzneimittel müssen bereits von der europäischen Zulassungsbehörde (EMA) oder den nationalen Zulassungsbehörden wie dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und dem Paul-Ehrlich-Institut (PEI) auf Qualität, Unbedenklichkeit und Wirksamkeit geprüft werden. Auf den ersten Blick mag es daher unverständlich erscheinen, warum ein zusätzliches Bewertungsverfahren, das HTA, durchgeführt werden soll. Doch Zulassung und HTA verfolgen jeweils unterschiedliche Ziele.
In der EU liegt es in der Verantwortung der einzelnen Mitgliedstaaten, eine flächendeckende Gesundheitsversorgung in ihrem Land sicherzustellen – entweder steuerfinanziert oder über ein Versicherungssystem. Da die Ressourcen in den Gesundheitssystemen begrenzt sind, stehen alle Länder vor derselben Herausforderung, zu entscheiden, welche Leistungen öffentlich finanziert werden und welche nicht. Hierfür wurde in vielen Ländern ein von der Zulassung zu unterscheidendes Bewertungsverfahren eingeführt, das andere Fragestellungen mit eigenen wissenschaftlichen Methoden behandelt.
In einem HTA wird systematisch geprüft, wie viel besser (oder schlechter) ein Arzneimittel, ein Medizinprodukt oder eine Therapie im Vergleich zu den besten verfügbaren Therapiealternativen für die Mehrzahl betroffener Patientinnen und Patienten ist. Es wird zudem beurteilt, ob die Ergebnisse der Studien auf die Versorgung im jeweiligen Land anwendbar sind und ob die positiven (oder negativen) Effekte für die Patientinnen und Patienten auch nach dem Ende der Studien bestehen bleiben. Das Ziel ist, Entscheidungen zur gerechten Verteilung der Ressourcen zu unterstützen. [/glossar]
[glossar] Warum gibt es ein EU - HTA Verfahren? :::
Seit fast 30 Jahren gibt es eine zentrale europäische Zulassung für Arzneimittel, doch ein gemeinsames HTA war bisher nicht vorgesehen. Der Grund dafür liegt in den unterschiedlichen Erstattungssystemen der einzelnen Länder, die sich unabhängig entwickelt haben.
In den meisten Ländern werden Kosten-Nutzen-Analysen durchgeführt. Die Kosteneffizienz der bewerteten Gesundheitstechnologie bestimmt maßgeblich die Erstattungsfähigkeit des Arzneimittels. In Deutschland hingegen sind verschreibungspflichtige Arzneimittel grundsätzlich erstattungsfähig, sobald sie auf den Markt kommen – mit wenigen Ausnahmen. In der so genannten „frühen Nutzenbewertung“ (AMNOG-Verfahren) wird vor allem der therapeutische Zusatznutzen eines Medikaments bewertet und dient als Grundlage für anschließende Preisverhandlungen. Ob ein Arzneimittel nach der Bewertung weiterhin für die Patientinnen und Patienten verfügbar ist, hängt davon ab, ob die Vertragsparteien sich auf einen Preis einigen können. Deutschland gehört im europaweiten Vergleich zu den Ländern, in denen die meisten zugelassenen neuen Arzneimittel auf dem Markt sind und die Patientinnen und Patienten am schnellsten Zugang zu diesen Arzneimitteln haben. Dennoch gibt es Arzneimittel, die in Deutschland nicht vermarktet werden und somit nicht zur Verfügung stehen.
Einige Länder verfügen über keine eigene HTA-Institution und nutzen daher keine HTA-Berichte für Erstattungsentscheidungen – sofern solche Entscheidungen überhaupt getroffen werden. Das bedeutet, dass der Zugang der Patientinnen und Patienten zu Arzneimitteln nicht allein von der Zulassung abhängt, sondern auch von der Bewertung durch HTA-Institutionen und den anschließenden Erstattungsentscheidungen.
In Ländern mit einem etablierten HTA-System mussten pharmazeutische Unternehmen bislang in jedem Land individuell die geforderten Nachweise entsprechend den nationalen Vorgaben aufbereiten und einreichen – dies in der jeweiligen Landessprache und zu unterschiedlichen Zeitpunkten. Mit dem EU-HTA Verfahren sollen diese Doppelarbeiten reduziert und Kompetenzen gebündelt werden.
Gemäß der Verordnung (EU) 2021/2282 über die Bewertung von Gesundheitstechnologien ist das Ziel der Regelungen vor allem:
- die Harmonisierung der Methoden zur Bewertung von Gesundheitstechnologien,
- die Schaffung von Transparenz,
- die Vermeidung von Redundanzen und
- einen schnelleren Zugang von Patientinnen und Patienten zu innovativen Therapien für ganz Europa herbeizuführen. [/glossar]
[glossar] Historisches zu EU HTA :::
Die erste formale HTA - Institution in Europa wurde 1987 in Schweden (Swedish Council on Technology Assessment in Health Care, SBU) gegründet. Nach und nach folgten andere europäische Länder, wie Spanien, Frankreich und das Vereinigte Königreich. In Deutschland wurde im Jahr 2000 zunächst das DIMDI (Deutsches Institut für Medizinische Dokumentation und Information) als Informationsinstitut für HTA gegründet, 2004 folgte das IQWiG (Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen) als Stiftung des Gemeinsamen Bundesausschusses (G-BA).
HTA – Institutionen der unterschiedlichen Länder haben über Jahrzehnte – in wechselnden Zusammensetzungen – in unterschiedlichen, zum Teil EU-geförderten Projekten freiwillig zusammengearbeitet. In der Zeit von 2021 bis 2023 wurden durch das Projekt - EUnetHTA 21 die maßgeblichen Vorbereitungen für Leitfäden und Prozesse erarbeitet, die die Grundlage für die nun folgende Epoche des europäischen HTA – Verfahrens bildet.
Als die EU – Kommission am 31. Januar 2018 einen ersten Entwurf einer HTA – Verordnung vorlegte, sahen Länder mit etabliertem HTA -System erhebliche Eingriffe in die rechtlich geschützte Zuständigkeit der Mitgliedstaaten für die Organisation ihrer Gesundheitswesen. Im März 2018 rügte beispielsweise der Deutsche Bundestag die Europäische Kommission wegen Verletzung der Grundsätze der Subsidiarität und der Verhältnismäßigkeit. Insbesondere wurden Qualitätseinbußen der in Deutschland etablierten frühen Nutzenbewertung befürchtet, die sieben Jahre zuvor mit dem Arzneimittelmarktneuordnungsgesetz (AMNOG) eingeführt wurde.
Nach intensivem Ringen zwischen der EU-Kommission, dem Europäischen Rat und dem Europäischen Parlament wurde die EU – HTA Verordnung verabschiedet und trat am 11. Januar 2022 in Kraft. Ab dem 12. Januar 2025 wird mit der Bewertung von Arzneimitteln für neuartige Therapien (ATMP) und onkologischen Arzneimitteln begonnen. In einem gestuften Verfahren werden ab 2028 Orphan Drugs und ab 2030 alle weiteren innovativen Arzneimittel folgen. [/glossar]
[glossar] EU-HTA: Organisationsstruktur :::
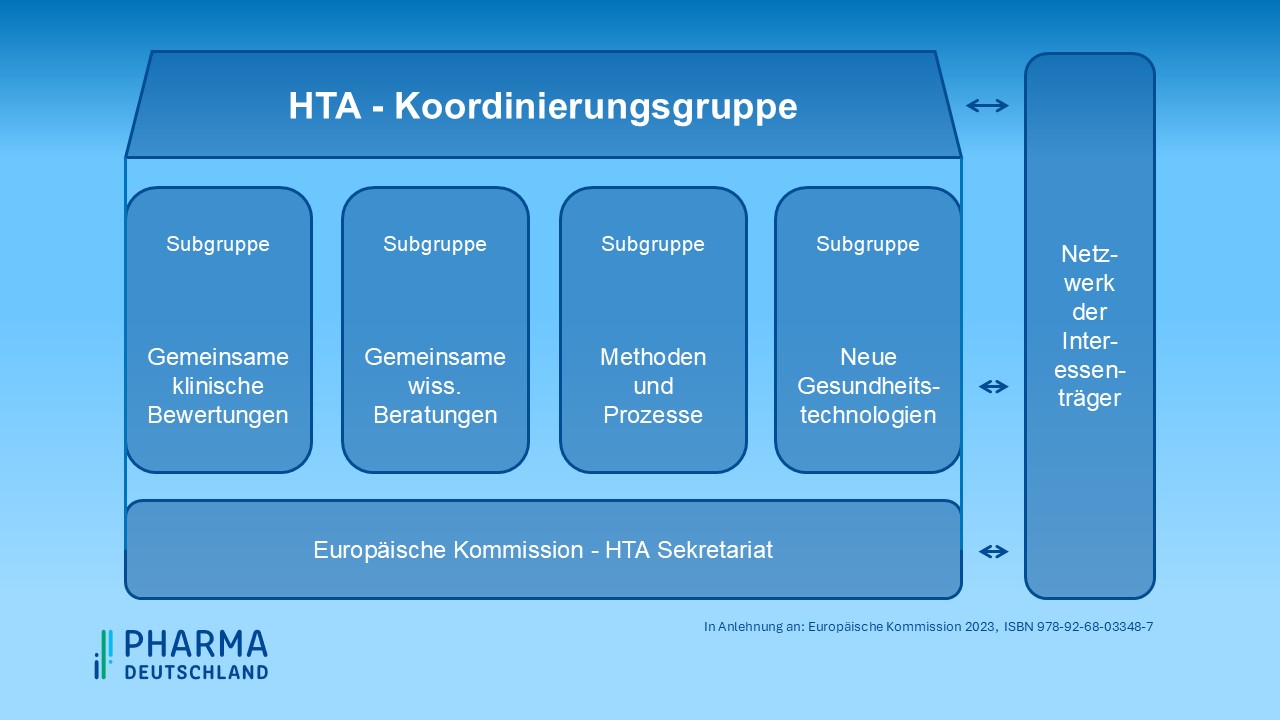
Die im März 2022 eingerichtete Koordinierungsgruppe leitet und überwacht die Arbeit der vier Subgruppen, die sich den Themenkomplexen Joint Clinical Assessment (gemeinsame klinische Bewertungen, JCA), Joint Scientific Consultations (gemeinsame wissenschaftliche Beratungen, JCA), Identification of emerging health technologies (neue Gesundheitstechnologien, EHT) und Methodological and procedural guidance (methodische und prozedurale Leitlinien, MPG) widmen. Unterstützt werden die Arbeiten von einem bei der Europäischen Kommission angesiedelten Sekretariat sowie dem im Juni 2023 etablierten Stakeholder-Netzwerk. Zudem wurde eine IT-Infrastruktur eingerichtet, die unter anderem dem Einreichen der Daten durch die Hersteller sowie der Veröffentlichung der HTA-Berichte dienen wird (siehe Abbildung 1).
Jeder Mitgliedstaat entsendet Vertreter sowohl in die Koordinierungsgruppe als auch in die Subgruppen. Die Vertreter müssen frei und unabhängig von Industrieinteressen sein. Auch wenn mehrere Vertreter entsendet werden, hat jeder Mitgliedstaat eine Stimme. Die Interessen Deutschlands werden durch Vertreter aus dem Bundesgesundheitsministerium für Gesundheit (BMG), des G-BA sowie dem IQWiG vertreten.
Dem Stakeholder-Netzwerk gehören Patientenvertreter, Nicht-Regierungsorganisationen im Bereich Gesundheit, Herstellerverbände sowie Klinische Experten an. Wesentlich ist, dass die Organisationen eine europaweite Organisation nachweisen können. Das Netzwerk wird in Stellungnahmeverfahren und in regelmäßige Informationsveranstaltungen eingebunden und unterstützt die Weiterentwicklung der Verfahren durch fachliche Expertise, Perspektiven und Feedback in verschiedenen Phasen der Bewertungen. Zur Beteiligung in diesem Netzwerk wird regelmäßig aufgerufen.
Abbildung 1: Verwaltungsstruktur in Anlehnung an Europäische Kommission 2023, ISBN 978-92-68-03348-7
[/glossar]
[glossar] Rechtliche Rahmenbedingungen, Leitfäden und weiterführende Links :::
EU-HTA Verordnung
In der Verordnung (EU) 2021/2282 über die Bewertung von Gesundheitstechnologien (EU-HTA Verordnung) werden die Eckpunkte der Verfahrensabläufe, die Aufgaben und Funktionen der zugeordneten Gremien sowie Rechte und Pflichten der Mitgliedsstaaten und der Hersteller der Gesundheitstechnologien (pharmazeutische Unternehmer und Medizinproduktehersteller) geregelt.
Durchführungsrechtsakte - Implementing Acts
In der EU-HTA Verordnung ist festgelegt, zu welchen Themenbereichen Durchführungsrechtsakte durch die Europäische Kommission zu erlassen sind. Diese sind rechtlich bindende Instrumente, die eingeführt werden, um die einheitliche Anwendung von Gesetzen oder Vorschriften sicherzustellen. Tabelle 1 zeigt eine Übersicht der Durchführungsrechtsakte.
Tabelle 1: Übersicht über die Durchführungsrechtsakte im Zusammenhang mit der EU-HTA Verordnung; Stand Januar 2025
| Thema | Kurzbeschreibung |
Gemeinsame klinische Bewertungen (JCA) (Mai 2024) | Verfahrensregeln und Fristen, Regeln für die Interaktion während des JCA für Arzneimittel, die Teilnahme an der Vorbereitung und Aktualisierung von JCAs |
Umgang mit Interessenkonflikten (25. Okt. 2024) | Regelungen zur Einholung, Prüfung und Umgang mit Interessenkonflikten für Vertreter der HTA – Koordinierungsgruppe, Untergruppen und Interessenvertretern wie Patienten und klinischen Experten |
Interaktion mit der Europäischen Arzneimittel-Agentur (18. Okt. 2024) | Planung und Prognose von JCA und JSC zur Auf-stellung des Jahresarbeits-programms und Planung der Fristen und Abläufe für die JCAs |
Gemeinsame wissenschaftliche Beratungen für Arzneimittel (18. Dez. 2024) | Verfahrensregeln für den Ablauf und für die Interaktion zwischen den Beteiligten inkl. Patienten und klinischen Experten im Rahmen eines JSC für Arzneimittel |
(24. Jan. 2024) | Verfahrensregeln für den Ablauf und für die Interaktion zwischen den Beteiligten inkl. Patienten und klinischen Experten im Rahmen eines JSC für Medizinprodukte |
| JCA für Medizinprodukte (ausstehend) | Verfahrensregeln und Fristen, Regeln für die Interaktion während des JCA für Medizinprodukte, die Teilnahme an der Vorbereitung und Aktualisierung von JCAs |
Richtlinien und Leitfäden
Richtlinien und Leitfäden unterstützen die Beteiligten bei der praktischen Umsetzung, Interpretation oder Konkretisierung rechtlicher Vorschriften und unterstützen die Konsistenz der Verfahren zu gewährleisten.
Die HTA – Koordinierungsgruppe hat zahlreiche methodische und praktische Richtlinien und Leitfäden verabschiedet, die sich größtenteils an die HTA – Gutachter und Entwickler von Gesundheitstechnologien richten. Eine Übersicht dieser Dokumente finden sich Tabelle 2 sowie weitere relevante Links in Tabelle 3.
Tabelle 2: Übersicht über die Richtlinien und Leitfäden im Zusammenhang mit der EU-HTA Verordnung; Stand Januar 2025
Thema Link zur Internetseite | Kurzbeschreibung Inhalt |
Anleitung für vornehmlich für Mitgliedsstaaten und Gutachter bzgl. der Definition und Formulierung von Endpunkten für den Scoping Prozess, mit Relevanz für Dossiererstellung | |
Umfasst die Definition, Klassifikation und Bewertung der Ergebnissicherheit von Studien | |
Anleitung für Gutachter zum Umgang mit Multiplizitäts-Problematiken und ergänzenden Analysen | |
Direct and indirect comparisons (methodische GL) | Beschreibung der verfügbaren Methoden für direkte und indirekte Vergleiche und method. Begriffsdefinitionen |
Direct and indirect comparisons , practical (praktische GL) | Anleitung für Gutachter zum Umgang mit den eingereichten Ergebnissen direkter und indirekter Vergleiche |
Zur Klarstellung der wissenschaftlichen Kriterien für einen „neuen Wirkstoff“ und für die Indikationszuordnung | |
Beschreibung der Verfahrensschritte für ein JCA | |
Beschreibung der Vorgehensweise bei der Auswahl und Bestimmung der Gutachter für JCA und JSC | |
Beschreibung des Vorgehens bei der Abfrage und Festlegung des Bewertungsumfanges für ein JCA | |
Umfasst mehrere Dokumente zur Erläuterung des Ausfüllens des EU-HTA Dossiers, inkl. Dossiervorlagen und separater Tabellenvorlagen | |
Beschreibung der Verfahrensschritte für ein JSC | |
Vorlage Beratungsantrag für JSC mit der EMA | |
Vorlage Beratungsantrag für JSC | |
Vorlage für Ergebnisniederschrift der Beratung | |
Beschreibung der Kriterien und Vorgehensweise für die Auswahl der Produkte für eine Beratung |
Weiterführende Links
Tabelle 3: weiterführende Links und Kontaktdaten
Relevante Internetlinks | |
Internetseite der Europäischen Kommission zu Health Technology Assessment | |
Internetseite der Europäischen Kommission zu gemeinsamen klinischen Bewertungen | |
Internetseite der Europäischen Kommission zu gemeinsamen wissenschaftlichen Beratungen | |
E-Mail-Kontakte |
|
Allgemein | |
JCA - produktspezifisch | |
JSC - produktspezifisch | |
für techn. Störungen der IT - Plattform | |
[/glossar]
[glossar] Ablauf des Verfahrens der gemeinsamen klinischen Bewertungen (JCA) :::
Unter die EU-HTA Verordnung fallen onkologische Arzneimittel mit neuen Wirkstoffen und ATMPs, die ab dem 12. Januar 2025 eine Zulassung beantragen. Nicht-onkologische Orphan – Drugs fallen erst ab dem 13. Januar 2028 und Nicht-onkologische Wirkstoffe erst ab dem 13. Januar 2030 unter die Regelung. Eine Hilfestellung zur Prüfung, ob der Wirkstoff unter die EU-HTA Verordnung fällt, gibt das Dokument „scientific specifications of medicinal products subject to joint clinical assessments“.
Der finale Bewertungsbericht über die Gesundheitstechnologie wird keine Werturteile zum Zusatznutzen der Arzneimittel enthalten, sondern ausschließlich die Evidenz und die Effekte der zu bewertenden Therapie gegenüber der jeweiligen Vergleichstherapie darstellen. Die einzelnen Mitgliedstaaten bleiben weiterhin verantwortlich, unter „gebührender Berücksichtigung“ der Ergebnisse des EU-HTA, auf nationaler Ebene Schlussfolgerungen zum klinischen Mehrwert zu ziehen. Um diesen Passus der Verbindlichkeit, bezogen auf die Verwendung der Ergebnisse in nationalen Verfahren, wurde bis zur Verabschiedung der EU-HTA Verordnung lange zwischen den politischen Lagern gerungen.
Der Ablauf des europäischen Nutzenbewertungsverfahrens für Arzneimittel gliedert sich in die folgenden fünf Phasen:
Initiierungsphase
- Information des HTA – Sekretariats über die Planung eines Zulassungsantrages, Versand des „Letter of Intent“ (Absichtserklärung einer Beantragung einer Zulassung) an die European Medicines Agency (EMA) und das HTA-Sekretariat inklusive aller erforderlichen Informationen
- Bestimmung der Gutachter seitens der HTA-Koordinierungsgruppe / Untergruppe und Identifikation von Patienten und klinischen Experten
- Veröffentlichung der Information über den Beginn des Verfahrens
Scoping Phase
- Einbindung der Patienten und klinischen Experten
- Übermittlung der Fragestellungen (PICO1) der Mitgliedsstaaten für das JCA
- Konsolidierung der Fragestellung
- Information des Herstellers über die im JCA zu beantwortenden Fragestellungen und Aufforderung zur Einreichung eines Dossiers
Dossiereinreichungsphase
- Erstellung des Dossiers anhand der Dossiervorgaben und methodischer Leitfäden
- Einreichung des Dossiers innerhalb der vorgegebenen Fristen (gemäß HTAR und Durchführungsrechtsakt für JCA mindestens 45 Tage vor der Positive Opinion des Ausschusses für Humanarzneimittel derEMA)
- Vollständigkeitsprüfung und Information über fehlende Informationen
- Ggf. Nachreichung fehlender Informationen
- Ggf. Aktualisierung des Dossiers im Falle von Anwendungsgebietsänderungen oder neuen Daten, die im Rahmen des Zulassungsprozesses bekannt werden
Bewertungsphase
- Bewertung des Dossiers durch die Gutachter
- Ggf. Rückfragen während der Bewertung an den pharmazeutischen Unternehmer
- Zusendung des HTA - Berichtentwurfes an den pharmazeutischen Unternehmer zum Zwecke der Faktenüberprüfung
- Abnahme des finalen HTA-Berichtes durch die HTA – Koordinierungsgruppe
Veröffentlichung
- Veröffentlichung des finalen HTA-Berichtes und des Dossiers ohne Betriebs- und Geschäftsgeheimnisse (Billigung des HTA-Berichts durch die Koordinierungsgruppe 30 Tage nach Entscheidung der EU-Kommission über die Zulassung + 10 Tage verfahrenstechnische Prüfung durch die EU-Kommission = 40 Tage nach Zulassung).
- Der Bericht darf keine Werturteile enthalten. Die Schlussfolgerungen, die die nationalen Gesundheitssysteme aus dem Bericht ziehen, obliegen weiterhin der Verantwortung der Mitgliedstaaten.
In Abbildung 2 sind die zeitlichen Fristen für ein reguläres Verfahren abgebildet. Bei beschleunigten Zulassungen oder Zulassungen neuer Anwendungsgebiete verkürzen sich die Zeitschienen entsprechend. Zudem kann sich die Veröffentlichung des Europäischen HTA – Berichtes inklusive des dazugehörigen Dossiers verzögern. Dies kann zu zeitlichen Konflikten mit dem Beginn des nationalen AMNOG – Verfahrens führen, denn derzeit ist mit Inverkehrbringen eines Arzneimittels mit neuem Wirkstoff in Deutschland die Verpflichtung verbunden, ein Dossier beim G-BA zum Zwecke der Nutzenbewertung einzureichen. Zu diesem Zeitpunkt beginnt das AMNOG – Verfahren und alle nachfolgenden Verfahrensschritte richten sich an diesem Startpunkt aus. Bei verzögerter Bereitstellung der EU-HTA Dokumente kann es daher zu Verschiebungen im AMNOG – Verfahren kommen.
Für detaillierte zeitliche Fristen und Verfahrensschritte des EU-HTA-Verfahrens sei auf die Procedural guidance for JCA medicinal products verwiesen.
[1] Die Fragestellung für die Bewertung definiert sich über die Patientenpopulation (im Anwendungsgebiet), die Intervention (das zu bewertende Arzneimittel), den Vergleich (Comparator) und die Endpunkte (Outcome). Diese Komponenten ergeben insgesamt den Bewertungsumfang = PICO.
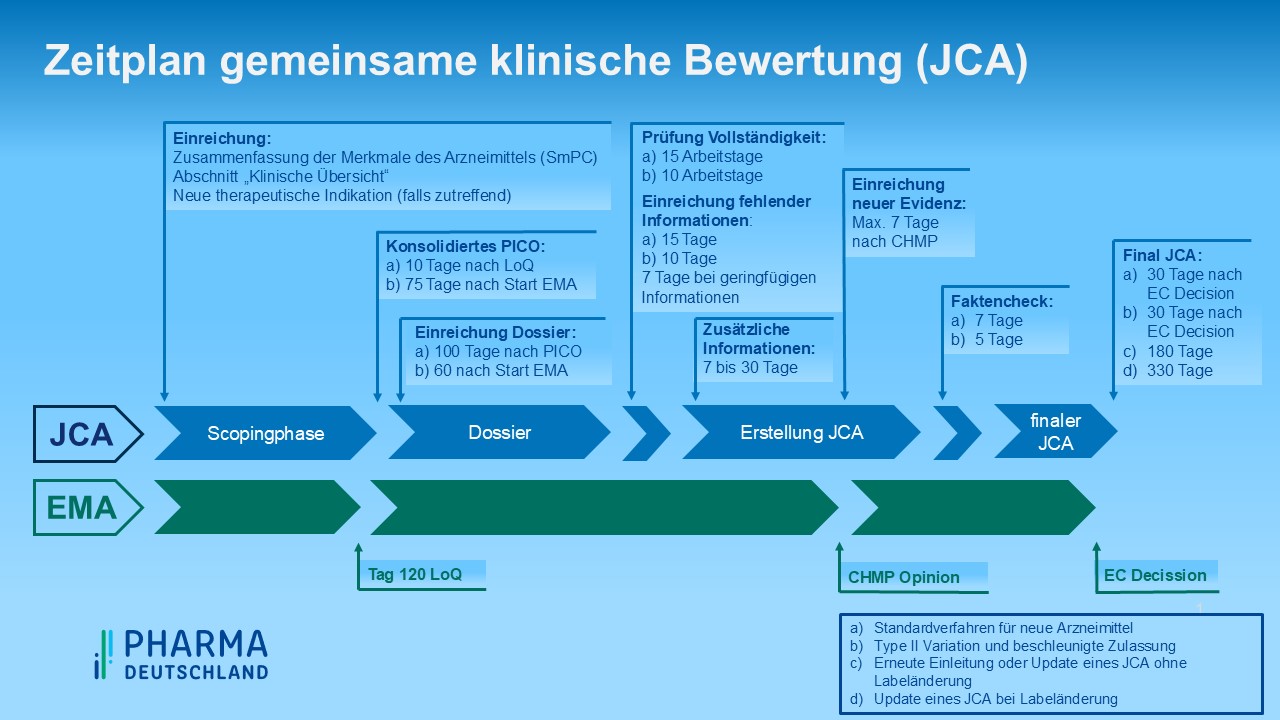
Abbildung 2: Zeitplan JCA, eigene Darstellung [/glossar]
[glossar] Verknüpfung mit den nationalen HTA – Verfahren :::
Das nationale Verfahren zur Nutzenbewertung verläuft bislang in mehreren Schritten: Zunächst reichen Unternehmen ein nationales Dossier gemäß der Modulvorlage des G-BA ein. Anschließend erstellt entweder das IQWiG oder der G-BA selbst (bei Orphan Drugs) ein Gutachten auf Basis der vorgelegten Daten. Die Ergebnisse werden auf der Website des G-BA veröffentlicht. Es folgt ein Stellungnahmeverfahren, das schriftlich und mündlich durchgeführt wird. Am Ende entscheidet das G-BA Plenum in einem Beschluss über den Zusatznutzen, der dann die Grundlage für die Erstattungsbetragsverhandlungen mit dem Spitzenverband Bund der Krankenkassen bildet.
Sowohl der EU-Bewertungsbericht als auch das EU-Dossier müssen in die nationale Bewertung einfließen. Da Unternehmen gemäß der EU-HTA Verordnung Unterlagen nicht erneut einreichen dürfen, sofern diese bereits auf EU-Ebene eingereicht wurden, muss bei der nationalen Bewertung auf die Ergebnisse der EU-HTA zurückgegriffen werden. Ergänzt werden darf nur, was national noch zusätzlich gefordert wird.
Um das europäische Verfahren mit dem nationalen Verfahren zu in Einklang zu bringen, hat das BMG am 2. Januar 2025 einen Referentenentwurf zur 1. Änderung der Verordnung über die Nutzenbewertung von Arzneimitteln zur Stellungnahme gestellt. Demnach kann der pharmazeutische Unternehmer nach Erteilung der Zulassung auch weiterhin den Zeitpunkt des Inverkehrbringens frei bestimmen und die grundlegenden Verfahrensschritte bleiben erhalten. Die Verpflichtung, ein Nutzenbewertungsdossier (mit Verweisen auf die Europäische Nutzenbewertung) beim G-BA einzureichen, besteht weiterhin. Der G-BA kann jedoch das Verfahren temporär bis zu 3 Monaten aussetzen, sofern die EU – HTA Dokumente bei Verfahrensstart noch nicht veröffentlicht worden sind. Dies kann beispielsweise der Fall sein bei Bewertungen von neuen Anwendungsgebieten, bei Verfahrensverzögerungen im EU-HTA-Prozess oder einer sehr schnellen Markteinführung des Arzneimittels in Deutschland. Ziel dieser Regelung ist es, ein reibungsloses Zusammenspiel der europäischen und nationalen Nutzenbewertung zu gewährleisten.
Wie genau die europäische Nutzenbewertung die deutsche Bewertungspraxis beeinflussen wird, bleibt abzuwarten. [/glossar]
[glossar] Was ändert sich für Patientinnen & Patienten und Ärztinnen & Ärzte? :::
Die Einführung des EU-HTA-Verfahrens ändert demnach weder den Zugang noch die Verordnungsfähigkeit neuer, innovativer Arzneimittel. Neue Medikamente bleiben auch weiterhin ab dem ersten Tag ihrer Markteinführung in Deutschland verordnungsfähig und der schnelle Zugang zu neuen Arzneimitteln für Patientinnen und Patienten gewährleistet.
Ärztinnen und Ärzte haben zudem die Möglichkeit, ihre klinische Expertise zusätzlich zum bereits etablierten Stellungnahmeverfahren im Rahmen der frühen Nutzenbewertung nun auch im EU-HTA-Verfahren einzubringen. Auf europäischer Ebene ist dies über europäisch organisierte Fachgesellschaften möglich. Darüber hinaus können bei spezifischen Produkten auch Einzelsachverständige beratend hinzugezogen werden. Dies gilt ebenfalls für Patientinnen und Patienten. Zu beachten ist, dass alle teilnehmenden Personen gemäß des entsprechenden Durchführungsrechtsaktes auf potenzielle Interessenkonflikte geprüft werden.
Im AMNOG-Stellungnahmeverfahren stehen neben den bekannten umfangreichen Dokumenten nun auch die Unterlagen aus dem europäischen HTA-Verfahren zur Verfügung. [/glossar]
[glossar] Wofür wir uns einsetzen :::
Trotz einer Vielzahl an veröffentlichten Richtlinien und Leitfäden, bleibt in der Interpretation und regelhaften Ausgestaltung der jetzt im Januar 2025 beginnenden neuen europäischen Nutzenbewertung noch viel Interpretationsspielraum. Insbesondere für pharmazeutische Unternehmen ist die praktische Umsetzung derzeit noch mit vielen Unsicherheiten verbunden. Bereits vor Beginn lassen sich Stolperstellen und Hürden für pharmazeutische Unternehmen identifizieren. Um die Europäische Nutzenbewertung zu einem Erfolg werden zu lassen, sind aus Sicht von Pharma Deutschland folgende Aspekte unerlässlich:
Hersteller sollten in der Lage sein, die Anforderungen im Rahmen des Bewertungsverfahrens mit angemessenem Aufwand zu bewältigen, das bedeutet:
- Begrenzte Anzahl an Fragestellungen im JCA: Die Anzahl der Fragestellungen nach dem PICO-Schema (Population, Intervention, Comparator, Outcome), sollte auf die relevanten, wesentlichen Fragestellungen begrenzt bleiben.
- Frühzeitige und faire Einbindung: Hersteller sollten für eine qualitativ hochwertige Dossiererstellung rechtzeitig über die geplanten Fragestellungen informiert und angemessen in deren Entwicklung einbezogen werden.
- Klare Vorgaben für die Nachweise: Sowohl auf europäischer Ebene als auch im nationalen Verfahren müssen eindeutige Vorgaben für die einzureichenden Nachweise bestehen, damit eine sorgfältige, aufwandsangemessene und planbare Vorbereitung der Dossiers möglich ist.
- Ausreichend Beratungsmöglichkeiten: Unternehmen sollten vor der Erstellung eines Dossiers und nicht nur während der Planung klinischer Studien Zugang zu ausreichenden Beratungsmöglichkeiten haben.
- Schutz von Betriebs- und Geschäftsgeheimnissen: Die Interessen der Unternehmen in Bezug auf Vertraulichkeit sensibler Daten müssen angemessen berücksichtigt werden.
- Keine Markteinführungsverzögerungen: Verfahren sollten so gestaltet sein, dass keine Verzögerungen bei der Markteinführung neuer Produkte entstehen.
Die Anforderungen an die Unternehmen müssen umsetzbar bleiben und gleichzeitig einen umfänglichen Informationsgewinn für das EU – HTA-Verfahren sicherstellen. Deshalb ist es essenziel, die Balance zwischen den Anforderungen an eine vollumfängliche Evidenzaufbereitung bei einer Vielzahl an Fragestellungen und dem Aufwand zur Aufbereitung der Daten zu gewährleisten. Zudem sollte die Nutzung und Verwendung der Bewertungsberichte in den Mitgliedsstaaten kontinuierlich evaluiert werden.
Pharma Deutschland informiert und berät seine Mitgliedsunternehmen umfangreich zu diesem Verfahren, um Wissen und Erfahrungen zu bündeln und gemeinsam das europäische Nutzenbewertungsverfahren auszugestalten. [/glossar]